Isom Lab News
 https://isomlabmichigan.com/wp-content/uploads/2025/04/IMG_4212-scaled.jpeg
2560
1920
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2025-04-22 17:05:322025-04-22 17:07:09Dr. Roberto Ramos-Mondragon becomes a US citizen!
https://isomlabmichigan.com/wp-content/uploads/2025/04/IMG_4212-scaled.jpeg
2560
1920
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2025-04-22 17:05:322025-04-22 17:07:09Dr. Roberto Ramos-Mondragon becomes a US citizen! https://isomlabmichigan.com/wp-content/uploads/2022/12/FFC6CBCF-CB10-4183-B82F-3246EAA79E3D_1_105_c.jpeg
1024
768
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2022-12-13 17:37:172022-12-13 17:41:37Dr. Isom elected to the National Academy of Medicine
https://isomlabmichigan.com/wp-content/uploads/2022/12/FFC6CBCF-CB10-4183-B82F-3246EAA79E3D_1_105_c.jpeg
1024
768
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2022-12-13 17:37:172022-12-13 17:41:37Dr. Isom elected to the National Academy of Medicine https://isomlabmichigan.com/wp-content/uploads/2022/12/EC89E2E4-8275-49AE-AE10-E4BF9D690B56_1_105_c.jpeg
768
1024
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2022-12-13 17:36:002022-12-13 17:39:03Dr. Isom receives the 2023 Basic Science Award from the American Epilepsy Society
https://isomlabmichigan.com/wp-content/uploads/2022/12/EC89E2E4-8275-49AE-AE10-E4BF9D690B56_1_105_c.jpeg
768
1024
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2022-12-13 17:36:002022-12-13 17:39:03Dr. Isom receives the 2023 Basic Science Award from the American Epilepsy Society https://isomlabmichigan.com/wp-content/uploads/2022/12/3A47F358-820F-4DBF-807E-976AF5578291_1_105_c.jpeg
768
1024
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2022-12-13 17:34:402022-12-13 17:50:21Isom Lab Celebrates at the 2023 AES Meeting in Nashville!
https://isomlabmichigan.com/wp-content/uploads/2022/12/3A47F358-820F-4DBF-807E-976AF5578291_1_105_c.jpeg
768
1024
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2022-12-13 17:34:402022-12-13 17:50:21Isom Lab Celebrates at the 2023 AES Meeting in Nashville! https://isomlabmichigan.com/wp-content/uploads/2021/07/Screen-Shot-2021-07-19-at-8.14.05-AM.png
1536
3088
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2021-07-19 12:17:252021-07-19 12:18:27Collaborative work by Veronica Beck with Dr. Anne Berg
https://isomlabmichigan.com/wp-content/uploads/2021/07/Screen-Shot-2021-07-19-at-8.14.05-AM.png
1536
3088
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2021-07-19 12:17:252021-07-19 12:18:27Collaborative work by Veronica Beck with Dr. Anne Berg https://isomlabmichigan.com/wp-content/uploads/2021/07/B72AE457-347D-4298-A0FE-6E40DFA6DF3B_1_105_c.jpeg
1024
768
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2021-07-14 19:40:272021-07-14 19:42:08Celebrating Together!
https://isomlabmichigan.com/wp-content/uploads/2021/07/B72AE457-347D-4298-A0FE-6E40DFA6DF3B_1_105_c.jpeg
1024
768
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2021-07-14 19:40:272021-07-14 19:42:08Celebrating Together! https://isomlabmichigan.com/wp-content/uploads/2017/08/Slide1.jpg
540
720
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2021-03-24 18:11:422021-03-24 18:11:42Open postdoctoral positions in the Isom Lab!
https://isomlabmichigan.com/wp-content/uploads/2017/08/Slide1.jpg
540
720
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2021-03-24 18:11:422021-03-24 18:11:42Open postdoctoral positions in the Isom Lab!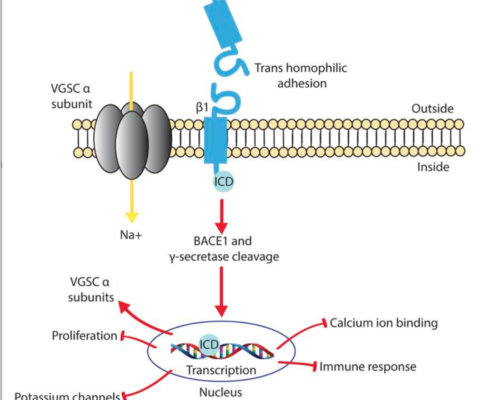 https://isomlabmichigan.com/wp-content/uploads/2021/03/Screen-Shot-2021-03-22-at-7.55.22-AM.png
1332
1518
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2021-03-22 11:55:542021-03-22 11:55:54Sodium channel β1 subunits participate in regulated intramembrane proteolysis-excitation coupling
https://isomlabmichigan.com/wp-content/uploads/2021/03/Screen-Shot-2021-03-22-at-7.55.22-AM.png
1332
1518
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2021-03-22 11:55:542021-03-22 11:55:54Sodium channel β1 subunits participate in regulated intramembrane proteolysis-excitation coupling https://isomlabmichigan.com/wp-content/uploads/2021/03/Screen-Shot-2021-03-22-at-7.37.55-AM.png
864
1518
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2021-03-22 11:39:322021-03-22 11:45:35Dancing to a different tune: TANGO gives hope for Dravet syndrome
https://isomlabmichigan.com/wp-content/uploads/2021/03/Screen-Shot-2021-03-22-at-7.37.55-AM.png
864
1518
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2021-03-22 11:39:322021-03-22 11:45:35Dancing to a different tune: TANGO gives hope for Dravet syndrome https://isomlabmichigan.com/wp-content/uploads/2021/01/Luis-crop.jpg
556
782
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2021-01-07 20:11:182021-01-07 20:13:24Mask up, sleeve up, Go Blue!
https://isomlabmichigan.com/wp-content/uploads/2021/01/Luis-crop.jpg
556
782
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2021-01-07 20:11:182021-01-07 20:13:24Mask up, sleeve up, Go Blue! https://isomlabmichigan.com/wp-content/uploads/2020/03/89056897_3124509234234094_44939307969937408_o.jpg
959
640
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2020-03-10 18:32:402020-03-10 18:34:32Congratulations to Dr. Allie Bouza!
https://isomlabmichigan.com/wp-content/uploads/2020/03/89056897_3124509234234094_44939307969937408_o.jpg
959
640
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2020-03-10 18:32:402020-03-10 18:34:32Congratulations to Dr. Allie Bouza! https://isomlabmichigan.com/wp-content/uploads/2019/08/IMG_0497-e1566482009306.jpg
3264
2448
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2019-10-31 18:17:112019-10-31 18:17:50Congratulations to Dr. Jacob Hull!
https://isomlabmichigan.com/wp-content/uploads/2019/08/IMG_0497-e1566482009306.jpg
3264
2448
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2019-10-31 18:17:112019-10-31 18:17:50Congratulations to Dr. Jacob Hull! https://isomlabmichigan.com/wp-content/uploads/2019/10/71074554_2750653788286309_93559944078426112_o.jpg
1269
1903
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2019-10-31 18:09:342019-10-31 18:19:04PIBS 20th Anniversary Celebration
https://isomlabmichigan.com/wp-content/uploads/2019/10/71074554_2750653788286309_93559944078426112_o.jpg
1269
1903
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2019-10-31 18:09:342019-10-31 18:19:04PIBS 20th Anniversary Celebration https://isomlabmichigan.com/wp-content/uploads/2019/10/70977792_2750653761619645_5660510009839058944_o.jpg
1296
1944
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2019-10-31 18:02:522019-10-31 18:08:11Congratulations to Allie Bouza
https://isomlabmichigan.com/wp-content/uploads/2019/10/70977792_2750653761619645_5660510009839058944_o.jpg
1296
1944
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2019-10-31 18:02:522019-10-31 18:08:11Congratulations to Allie Bouza https://isomlabmichigan.com/wp-content/uploads/2018/10/2018-10-16_14-19-51-1.jpg
535
535
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2018-10-16 21:18:212018-10-20 22:31:41Basic Science 2018
https://isomlabmichigan.com/wp-content/uploads/2018/10/2018-10-16_14-19-51-1.jpg
535
535
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2018-10-16 21:18:212018-10-20 22:31:41Basic Science 2018 https://isomlabmichigan.com/wp-content/uploads/2018/08/Jacob-Veronica-2018-GRC.jpg
2160
2880
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2018-08-27 21:52:192018-08-27 21:52:51Mechanisms of Epilepsy and Neuronal Synchronization GRS/GRC at Mt. Snow, VT
https://isomlabmichigan.com/wp-content/uploads/2018/08/Jacob-Veronica-2018-GRC.jpg
2160
2880
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2018-08-27 21:52:192018-08-27 21:52:51Mechanisms of Epilepsy and Neuronal Synchronization GRS/GRC at Mt. Snow, VT https://isomlabmichigan.com/wp-content/uploads/2018/08/stemcr-11-3-mockup.jpg
3438
2725
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2018-08-27 20:38:372023-06-05 09:46:07We got the cover!
https://isomlabmichigan.com/wp-content/uploads/2018/08/stemcr-11-3-mockup.jpg
3438
2725
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2018-08-27 20:38:372023-06-05 09:46:07We got the cover! https://isomlabmichigan.com/wp-content/uploads/2018/04/Nnamdi-poster-award-PSTP-2018.jpg
1365
2048
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2018-04-27 21:01:092018-04-27 21:01:09Congratulations to Nnamdi Edokobi on winning the runner-up best poster award at the 2018 PSTP Symposium
https://isomlabmichigan.com/wp-content/uploads/2018/04/Nnamdi-poster-award-PSTP-2018.jpg
1365
2048
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2018-04-27 21:01:092018-04-27 21:01:09Congratulations to Nnamdi Edokobi on winning the runner-up best poster award at the 2018 PSTP Symposium https://isomlabmichigan.com/wp-content/uploads/2018/04/Allie-poster-award-PSTP-2018.jpg
1365
2048
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2018-04-27 20:56:452018-04-27 20:58:58Congratulations to Allie Bouza on winning the best poster award at the 2018 PSTP Symposium
https://isomlabmichigan.com/wp-content/uploads/2018/04/Allie-poster-award-PSTP-2018.jpg
1365
2048
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2018-04-27 20:56:452018-04-27 20:58:58Congratulations to Allie Bouza on winning the best poster award at the 2018 PSTP Symposium https://isomlabmichigan.com/wp-content/uploads/2018/04/Allie-Diversity-Grant-Award.jpg
1512
2016
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2018-04-10 01:05:592023-02-22 11:54:44Congratulations to Allie Bouza 2018 Diversity Equity and Inclusion Grant Recipient
https://isomlabmichigan.com/wp-content/uploads/2018/04/Allie-Diversity-Grant-Award.jpg
1512
2016
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2018-04-10 01:05:592023-02-22 11:54:44Congratulations to Allie Bouza 2018 Diversity Equity and Inclusion Grant Recipient https://isomlabmichigan.com/wp-content/uploads/2017/08/team-allie.jpg
450
450
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2018-03-13 21:07:332018-04-27 21:10:29Allie Bouza receives an impact score of 10 and percentile score of 1 on her NIH-NRSA F31 predoctoral fellowship.
https://isomlabmichigan.com/wp-content/uploads/2017/08/team-allie.jpg
450
450
Lori Isom
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
Lori Isom2018-03-13 21:07:332018-04-27 21:10:29Allie Bouza receives an impact score of 10 and percentile score of 1 on her NIH-NRSA F31 predoctoral fellowship.  https://isomlabmichigan.com/wp-content/uploads/2017/12/20988905_10210694656738920_7731692544747120414_o.jpg
1365
2048
jdmcmah
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
jdmcmah2017-08-20 23:09:222017-12-10 00:07:23Congratulations to Lori Isom – appointed as the third Maurice H. Seevers Professor
https://isomlabmichigan.com/wp-content/uploads/2017/12/20988905_10210694656738920_7731692544747120414_o.jpg
1365
2048
jdmcmah
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
jdmcmah2017-08-20 23:09:222017-12-10 00:07:23Congratulations to Lori Isom – appointed as the third Maurice H. Seevers Professor https://isomlabmichigan.com/wp-content/uploads/2017/08/alli-teaches-wolverine-pathways.jpg
450
450
jdmcmah
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
jdmcmah2017-08-19 19:15:542017-08-19 19:19:31Allie Bouza leads Wolverine Pathways summer program in Pharmacology
https://isomlabmichigan.com/wp-content/uploads/2017/08/alli-teaches-wolverine-pathways.jpg
450
450
jdmcmah
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
jdmcmah2017-08-19 19:15:542017-08-19 19:19:31Allie Bouza leads Wolverine Pathways summer program in Pharmacology https://isomlabmichigan.com/wp-content/uploads/2017/08/chad.jpg
450
450
jdmcmah
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
jdmcmah2017-08-19 18:52:392024-02-20 12:40:09Congratulations to Chad Frasier, PhD (Isom Lab), 2017 AES Early Career Award Recipient
https://isomlabmichigan.com/wp-content/uploads/2017/08/chad.jpg
450
450
jdmcmah
http://isomlabmichigan.com/wp-content/uploads/2017/08/logo-300x42.png
jdmcmah2017-08-19 18:52:392024-02-20 12:40:09Congratulations to Chad Frasier, PhD (Isom Lab), 2017 AES Early Career Award Recipient